Alzheimer’s disease via enhanced calcium signaling caused by the decrease of endoplasmic reticulum–mitochondrial distance
Abstract
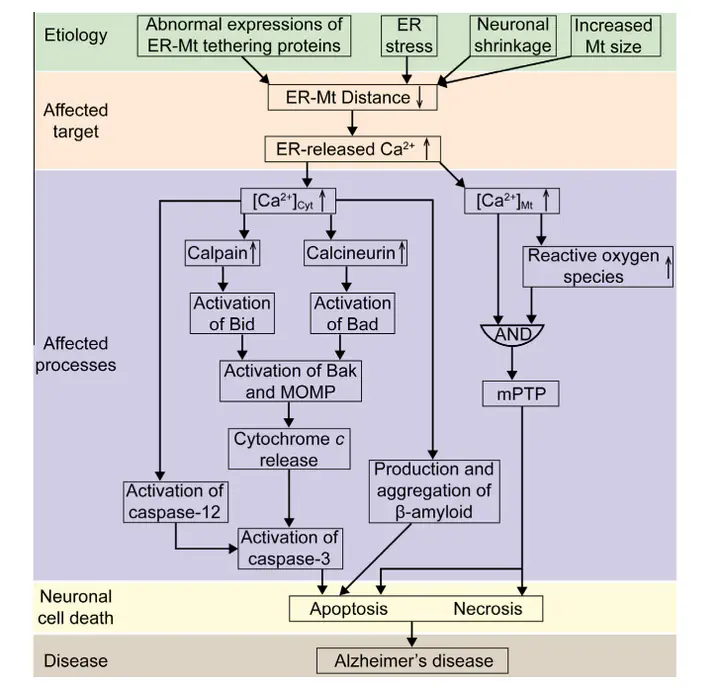
It has long been recognized that Ca(2+) dysregulation is relevant to the initiation of Alzheimer’s disease (AD), and most recent works have suggested that increased cross-talk between endoplasmic reticulum (ER) and mitochondria plays an important role in the pathogenesis of the disease. However, the detailed mechanism involved has not been fully elucidated. Owing to its importance in the regulation of Ca(2+) signaling, ER-mitochondrial distance in the neurons is tightly controlled in the physiological conditions. When the distance is decreased, Ca(2+) overload occurs both in the cytosol and mitochondria. The cytosolic Ca(2+) overload can (1) hyperactivate Ca(2+)-dependent enzymes, which in turn regulate activities of pro-apoptotic BCL-2 family proteins, causing mitochondrial outer membrane permeabilization and thereby resulting in the release of cytochrome c to activate caspase-3; (2) indirectly activate caspase-3 through the activation of caspase-12; and (3) promote the production and aggregation of β-amyloid. The three pathways eventually trigger neuronal apoptotic cell death. The mitochondrial Ca(2+) overload can lead to increased generation of reactive oxygen species, inducing the opening of the mitochondrial permeability transition pore and ultimately causing neuronal apoptotic and necrotic cell death. The resultant death of neurons which are responsible for memory and cognition would contribute to the pathogenesis of AD. Therefore, we propose that the reduction in the distance between ER and mitochondria may be implicated in AD pathology by enhanced Ca(2+) signaling, which provides a more complete picture of the Ca(2+) hypothesis of AD