MSSort-DIA XMBD: A deep learning classification tool of the peptide precursors quantified by OpenSWATH
Abstract
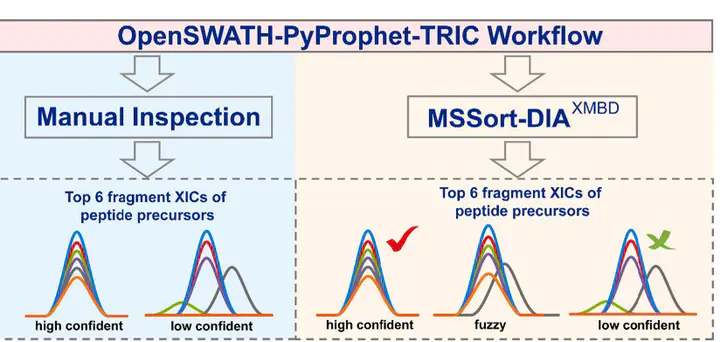
OpenSWATH is an analysis toolkit commonly used for data independent acquisition (DIA).
Although the output of OpenSWATH is controlled at 1% false discovery rate (FDR), the output report still
contains many peptide precursors with low similarity fragments. At the last step of OpenSWATH for peptide
quantification, researchers usually need to manually check the similarity of the extracted ion chromatograms
(XICs) of fragments to distinguish the high confidence and the low confidence peptide precursors. Here we
developed an algorithm with a Graphic User Interface named MSSort-DIAXMBD, which combines the deep
convolutional neural network (CNN) and the double-threshold segmentation process, to automatically recognize
the high confidence precursors and low confidence precursors. To train the model of MSSort-DIAXMBD, we built a
database contained about 50,000 manually classified peptide precursors acquired from different instrument
platforms and different species. With the double-threshold segmentation strategy, MSSort-DIAXMBD can reduce
the number of the low confidence peptides required for manual inspections to less than 10% and be used as the
last step of OpenSWATH to visualize and classify the MS/MS data of peptide precursors.
SIGNIFICANCE: Although the output of OpenSWATH is controlled at 1% false discovery rate
(FDR), the output report still contains many peptide precursors with low similarity fragments. At the last
step of OpenSWATH for peptide quantification, researchers usually need to manually check the similarity of
fragment XICs to distinguish the high confidence and the low confidence peptide precursors. However, manual
inspection is inefficient. For instance, it takes about 50 h to sort even a small dataset of 1000 MS/MS
spectra manually. In this paper we developed a software named MSSort-DIAXMBD to automatically recognize the
high confidence precursors. We manually classify 50,000 peptide precursors as training set to train a
convolutional neural network. After training finished, MSSort-DIAXMBD takes only a few minutes to
automatically classify 20,000 peptide precursors, leaving a small portion of fuzzy ones for manual inspection.
On the benchmarked dataset, MSSort-DIAXMBD can significantly improve the efficiency and accuracy of
recognition of high confidence peptide precursors.
Keywords: Data-independent
acquisition proteomics; Deep convolutional neural networks; Deep learning; OpenSWATH.